What causes protein misfolding diseases?

The processes that govern life within our cells are miracles of microscopic engineering, relying on countless molecules to fold into precise three-dimensional shapes. When this fundamental requirement is broken—when a protein adopts the wrong shape—the consequences can cascade into devastating, often incurable, diseases. This phenomenon, where proteins aggregate into toxic forms, is the underpinning cause of a group of disorders collectively termed proteinopathies. [7] Understanding what triggers this initial folding error is key to understanding conditions that affect millions, ranging from Alzheimer's and Parkinson's diseases to Creutzfeldt-Jakob disease. [2][3]
# Shape Function
Proteins are synthesized as linear chains of amino acids, but their function is entirely dependent on their final, unique architecture. This intricate folding process is rapid and highly specific, driven by chemical forces that dictate which parts of the chain interact with others, forming secondary structures like alpha-helices and beta-sheets, which then assemble into the final functional tertiary or quaternary structure. [4] A protein must attain its native conformation to perform its role, whether as an enzyme catalyzing a reaction, a structural component maintaining cell shape, or a signaling molecule transmitting information. [1]
When a protein fails to achieve this correct native state, or when a correctly folded protein undergoes an unexpected structural change later in life, it is termed a misfolded protein. [1] This error introduces "sticky" surfaces that were never meant to be exposed. These exposed hydrophobic regions or altered surfaces cause the misfolded proteins to stick together, initiating aggregation rather than performing their intended job. [4]
# Misfolding Triggers
The deviation from the native state can stem from various points in a protein's life cycle, making the cause of a specific proteinopathy often multifactorial. A primary driver is simply genetic mutation. [1] A change in the DNA code can alter a single amino acid within the protein chain. While some mutations are silent, others can dramatically change the folding landscape, favoring an alternative, pathogenic shape. [4] For example, in familial amyloidosis, a specific point mutation in the transthyretin protein makes it highly prone to misfolding and fibril formation. [1]
Beyond inherent genetic flaws, the cellular environment plays a crucial, dynamic role. Stressors that compromise the cell’s ability to maintain proper conditions—such as oxidative stress, inflammation, or mitochondrial dysfunction—can destabilize protein structures. [9] If the cellular machinery responsible for managing protein health is overwhelmed or damaged, even a perfectly coded protein can begin to drift toward misfolding. [1]
Consider the cumulative effect of cellular age. As a cell ages, its internal environment shifts; the concentration of beneficial stabilizing ions might decrease, or the overall chemical balance might become less ideal for protein stability. This means that the same protein sequence that folds perfectly at age 20 may become marginally less stable at age 70, tipping the balance toward misfolding simply due to accumulated minor environmental insults over decades. [4]
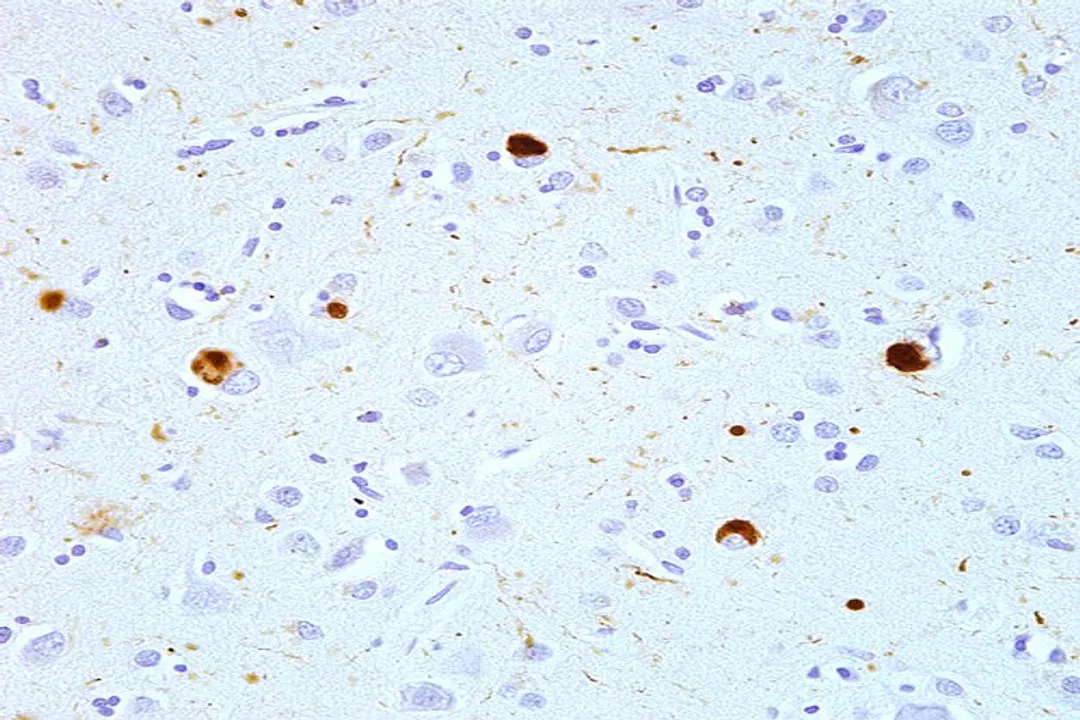
# Toxic Intermediates
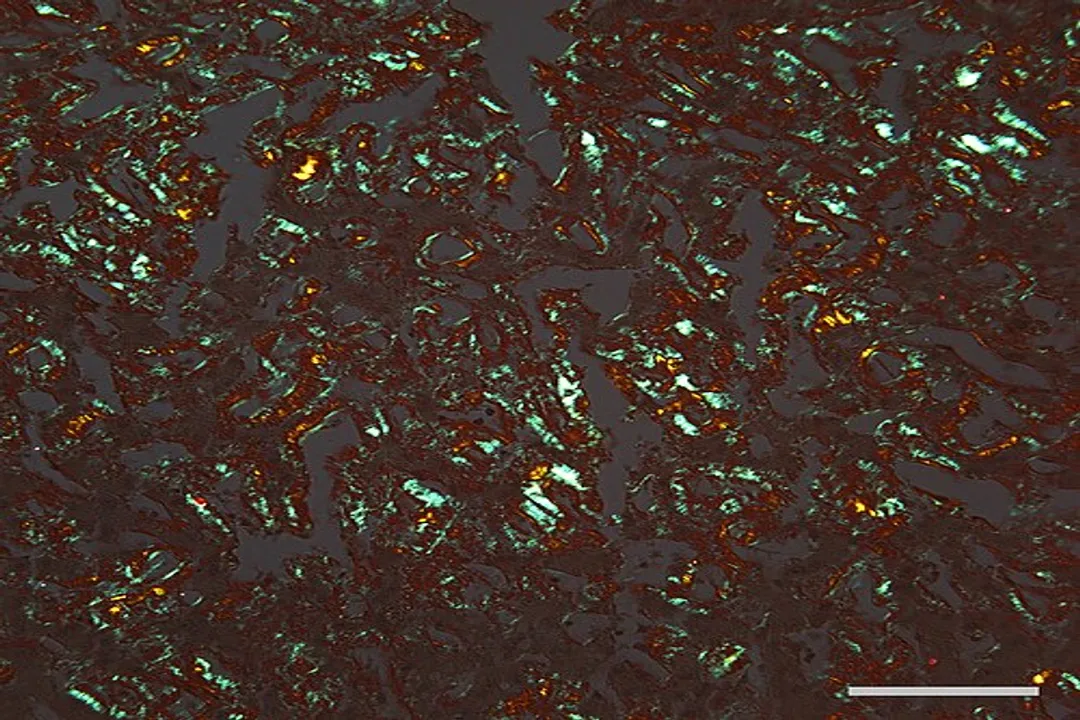
It is a common misconception that the massive, insoluble plaques or fibrils seen in diseases like Alzheimer's are the primary toxic agents. While these large aggregates are hallmarks of the condition, the most damaging species appear to be the soluble oligomers. [4][1] These are intermediate structures—small clusters of two to perhaps twenty misfolded proteins—that form before the final, large amyloid fibrils. [3]
These oligomers are particularly disruptive because they can interact with and impair essential cellular components. They are known to bind to and disrupt synaptic function at the neuronal membrane, interfere with mitochondrial energy production, and impede the function of the proteasome, the cell's primary protein disposal system. [2][4]
The progression often follows a pattern: a monomer (single protein unit) misfolds, joins a small, highly toxic oligomer, and eventually, this structure co-opts more monomers, growing into an insoluble, less reactive fibril deposit. [3] While the long-term presence of these large deposits causes physical disruption and triggers inflammation, the acute toxicity that drives the early symptoms is often attributed to the smaller, transient oligomeric species. [4]
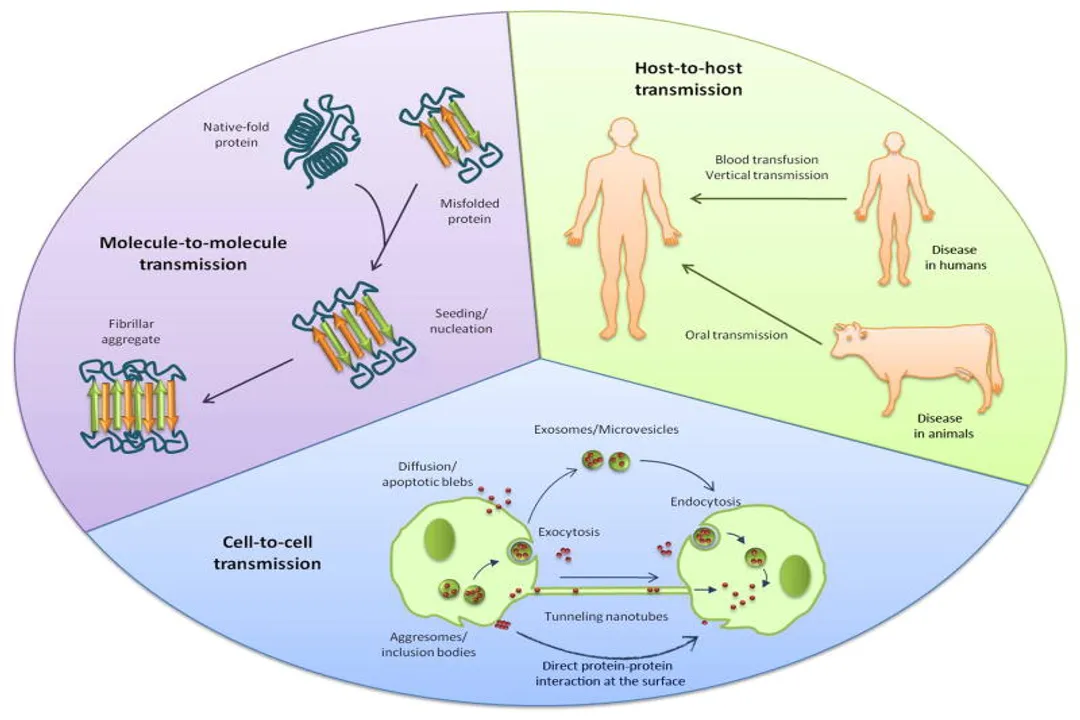
# Propagation Mechanisms
A particularly concerning aspect of these diseases is their ability to spread, moving from affected cells to healthy ones. This is where the concept of template-directed misfolding becomes critical, most famously illustrated by prions. [6] Prions are infectious protein agents where the misfolded protein structure acts as a template to convert correctly folded native proteins into the pathogenic conformation. [8]
The process, often described in the context of neurodegenerative disease, involves a seeding mechanism. A misfolded protein (the seed) interacts with a normally folded protein, forcing it to adopt the pathological conformation, thus creating two misfolded proteins where there was one before. [6] This conversion mechanism is not limited to true prions (like those causing Bovine Spongiform Encephalopathy) but is now understood to apply to the pathological forms of proteins like alpha-synuclein in Parkinson's disease and amyloid-beta/tau in Alzheimer's disease. [6]
This templating effect allows the disease state to propagate through tissue, often following anatomical pathways, much like an infection spreading through the nervous system. [6] For instance, alpha-synuclein pathology in Parkinson's disease appears to start in the enteric nervous system or the lower brainstem and progressively move upward into the higher brain centers, mirroring the progression of motor symptoms. [2] The efficiency of this seeding dictates how rapidly the pathology develops and spreads within the organism. We can conceptually categorize the risk of propagation based on the protein's inherent propensity to seed, which is often linked to the stability of its resulting fibril structure. A protein that forms a highly stable, rigid seed, like the scrapie form of PrP, will propagate aggressively, whereas others might require more significant external cellular distress to overcome the energy barrier required for initial seeding. [8]
# Cellular Quality Control Failure
The cell has evolved elaborate systems to prevent and correct misfolding. This protein homeostasis (proteostasis) network relies on molecular chaperones, which are specialized proteins that bind to nascent or stressed proteins, helping them fold correctly or keeping them soluble until conditions improve. [1][5] When chaperones cannot rescue a misfolded protein, the cell targets it for destruction, primarily through the ubiquitin-proteasome system (UPS) or by packaging it into autophagosomes for degradation via lysosomes. [5]
Protein misfolding diseases arise, therefore, not just because a protein can misfold, but because the quality control machinery has either failed, been overwhelmed, or been directly inhibited by the misfolded species themselves. [1]
For example, the accumulation of large amyloid deposits can physically clog the machinery of the proteasome, creating a feedback loop where the disposal system becomes less effective precisely when it is needed most. [5] Furthermore, the cellular machinery, including chaperones, can become depleted or functionally compromised due to age or chronic stress, shifting the entire balance towards aggregation. [9]
| Quality Control Component | Primary Function | Failure Consequence |
|---|---|---|
| Chaperones | Assist correct folding, prevent premature aggregation [1] | Increased population of soluble, toxic oligomers [5] |
| Proteasome (UPS) | Tag and degrade short-lived or damaged proteins [5] | Buildup of ubiquitinated/misfolded proteins in the cytoplasm |
| Autophagy/Lysosomes | Degrade large aggregates and damaged organelles [5] | Formation of large, sometimes resistant, intracellular inclusions |
The failure of this network highlights a crucial point: these diseases are often diseases of aging and system failure rather than just diseases of protein structure alone. [9] A healthy, young cell can often sequester or clear small amounts of misfolded intermediates that might overwhelm a stressed, older cell.
# Major Pathological Proteins
The spectrum of diseases linked to protein misfolding—proteinopathies—is broad, defined by which specific protein is involved and where it accumulates in the body. [7]
In Alzheimer's disease (AD), two proteins are centrally implicated: Amyloid-beta () and Tau. is thought to aggregate outside the neurons, forming the characteristic extracellular plaques. Tau protein, normally stabilizing microtubules inside the neuron, becomes hyperphosphorylated, detaches, and forms intracellular neurofibrillary tangles. [3]
Parkinson's disease (PD) is characterized by the accumulation of alpha-synuclein within neurons, forming Lewy bodies. [2] These aggregates are toxic to dopaminergic neurons, leading to the classic motor symptoms. Research continues on new ways to target this pathology, perhaps by modulating the seeding process or enhancing clearance mechanisms specifically adapted to alpha-synuclein's structure. [10]
Prion diseases (like CJD) are unique because the misfolded protein, , is capable of transmission between individuals (or animals) through ingestion or contact, acting as a highly potent infectious agent via its template-directed conversion activity. [7][8] Other diseases involve proteins like transthyretin (amyloidosis) or Huntingtin (Huntington's disease). [1]
# Environmental and Therapeutic Angles
While genetics sets the stage, external and lifestyle factors often determine when, or if, the disease manifests. Factors that increase protein turnover or demand more efficient folding place stress on the system. Poor sleep, for example, has been linked to impaired clearance of from the brain during the night, suggesting that basic physiological maintenance is intrinsically tied to preventing proteotoxicity. [2]
From a therapeutic perspective, understanding the cause of the misfolding guides the approach. One strategy focuses on preventing the initial error, perhaps through gene therapy to correct a mutation or through small molecules that stabilize the native fold (a concept known as pharmacological chaperoning). [1] Another, more common approach is to interrupt the aggregation process once it has started. This involves designing molecules that either bind to the toxic oligomers, preventing them from interacting with the cell membrane, or blocking the seeding interface, thereby halting the template-driven conversion. [10]
The challenge in developing effective treatments lies in intervening at the right time. If intervention occurs only after significant fibril deposition has already happened, the damage might be irreversible. If the intervention is too early, it may be unnecessary or expose the patient to unwarranted side effects from blocking a normal physiological process. [4] A deep knowledge of the kinetics—how fast the native protein converts to oligomer, and how fast the oligomer converts to stable fibril—is essential for effective drug timing. The difference in pathology between a highly infectious prion seed and a slowly accumulating seed dictates that no single therapeutic strategy will likely work for all proteinopathies; the solution must be specific to the structural characteristics of the misfolded culprit. [8] This specificity is what makes research in this area so complex yet so necessary for combating these challenging conditions.
Related Questions
#Citations
Misfolded Protein Aggregates: Mechanisms, Structures and ...
Misfolded proteins and neurodegenerative diseases | BMG LABTECH
Protein Misfolding and Degenerative Diseases - Nature
Protein Misfolding - an overview | ScienceDirect Topics
Protein-misfolding diseases and chaperone-based therapeutic ...
Protein misfolding that propagates and the mechanisms of ...
Proteinopathy - Wikipedia
How do prions misfold other proteins? : r/askscience - Reddit
Molecular and cellular aspects of protein misfolding and disease
A New Approach to Protein Misfolding in Parkinson Disease