How do prions cause disease?
The fundamental strangeness of prion diseases lies in their causative agents: prions are infectious entities composed almost entirely of protein, a concept that challenges the long-held central dogma of microbiology which posits that infectious agents must contain nucleic acids like DNA or RNA. [4][7] These agents are responsible for a group of fatal neurodegenerative disorders characterized by the progressive loss of brain cells and the development of microscopic, sponge-like holes in the affected tissue. [1][3][8] Understanding how these rogue proteins wreak havoc requires looking closely at their structure and their sinister ability to coerce normal biological molecules into adopting their destructive shape. [7]
# Protein Identity
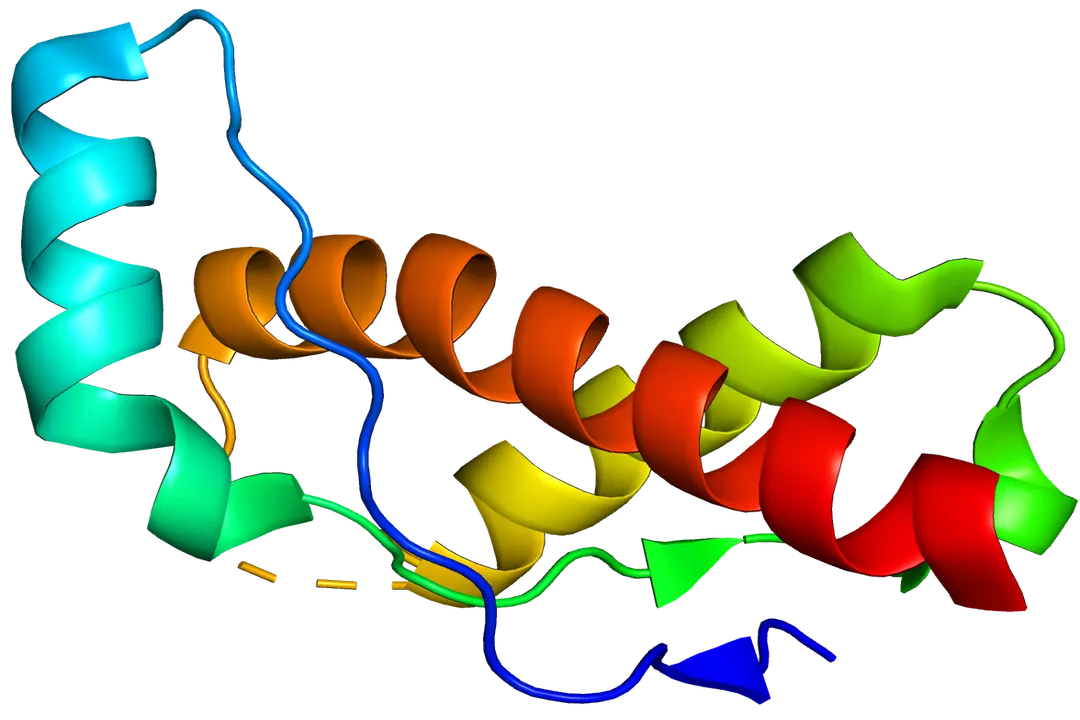
The mystery begins with a perfectly normal, indispensable protein called the cellular prion protein, denoted as . [1][4] This standard protein is found anchored to the outer surface of many cells throughout the body, though it is most abundant in the nervous system, particularly on the membranes of neurons. [1][4] In its healthy configuration, is soluble and has a structure rich in what scientists call alpha-helices. [5] Its exact physiological function is still under investigation, but it is thought to be involved in cell signaling, copper metabolism, and possibly protecting cells from oxidative stress. [7]
The trouble starts when this benign protein encounters its pathogenic twin, the scrapie form, designated . [1][7] The difference between and is not in their amino acid sequence—they are made of the same building blocks—but in their three-dimensional folding pattern. [4][5] Where the normal protein relies on soft, spiraling alpha-helices, the infectious version adopts a configuration dominated by rigid, flat structures known as beta-sheets. [5] This seemingly minor change in architecture transforms a helpful cellular component into a highly resistant, toxic agent. [4] The name "prion" itself is a contraction derived from "proteinaceous infectious only". [4]
# Conversion Mechanism
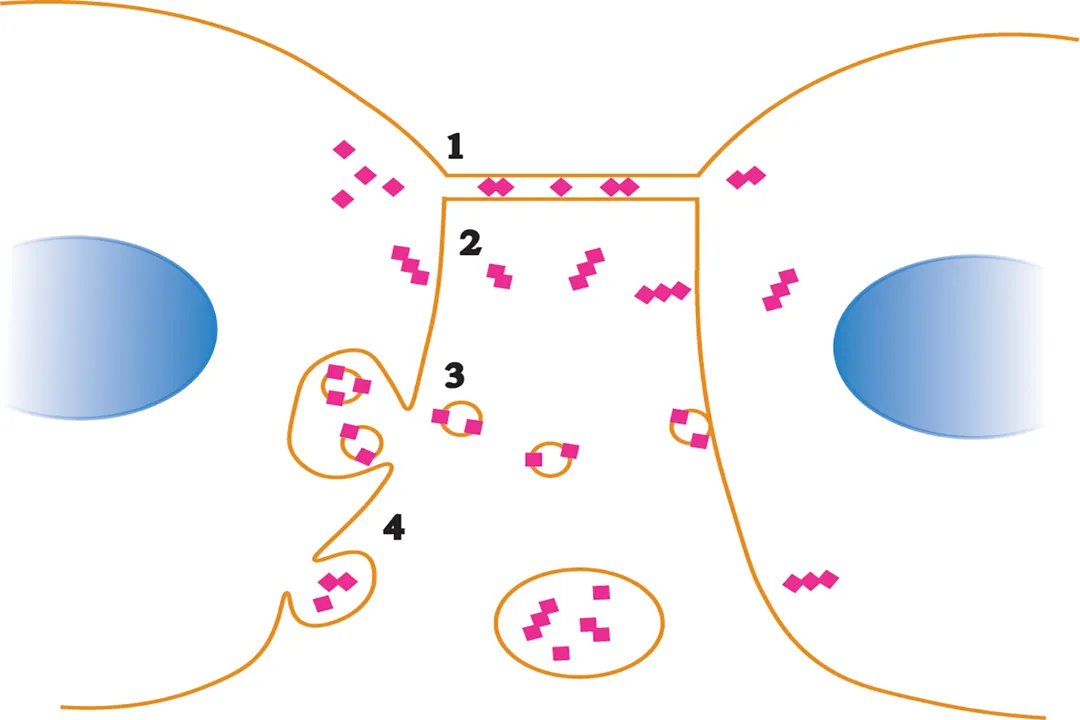
The mechanism by which prion diseases propagate is a self-sustaining chain reaction, akin to a domino effect initiated by the abnormal protein. [7] When a normal molecule physically interacts with an invading or newly formed molecule, the abnormal protein acts as a highly effective template. [1][4][7] This template forces the native to refold, abandoning its native, alpha-helical structure and adopting the infectious, beta-sheet-rich conformation of . [1][7]
This conversion process is the heart of prion infectivity. [5] Each newly converted molecule is then capable of recruiting and converting yet another surrounding , causing the misfolded population to grow exponentially. [7] This autocatalytic conversion means that once the process begins, the concentration of the toxic agent increases rapidly, even if the initial infectious dose was small. [4] Unlike viruses, which must hijack the cell's machinery to replicate their genetic material, prions propagate by direct conformational change, making them entirely self-propagating protein assemblies. [7]

# Aggregation Pathway
The danger escalates once the conversion is complete because the resulting proteins exhibit drastically different physical properties than their cellular counterparts. [4] The infectious isoforms are notoriously stable, relatively insoluble, and highly resistant to breakdown by the cell’s normal waste disposal systems, specifically proteases, the enzymes designed to break down damaged or misfolded proteins. [1][4][7] This resistance to degradation is a critical factor in the pathology, allowing these aberrant proteins to persist and accumulate within the host tissue for long periods. [4]
As more and more molecules are generated, they begin to associate with each other, clumping together in a process known as aggregation. [1][4] These aggregates can form long, fibrous structures called amyloid fibrils, which further coalesce into larger, insoluble deposits known as amyloid plaques within the brain parenchyma. [5] The sheer physical presence of these growing deposits disrupts the normal cellular environment, contributing to toxicity and eventual cell death. [1] One way this disruption manifests is by sequestering or interfering with the function of normal, essential cellular components, essentially starving the neurons of necessary molecules. [5]
# Tissue Destruction
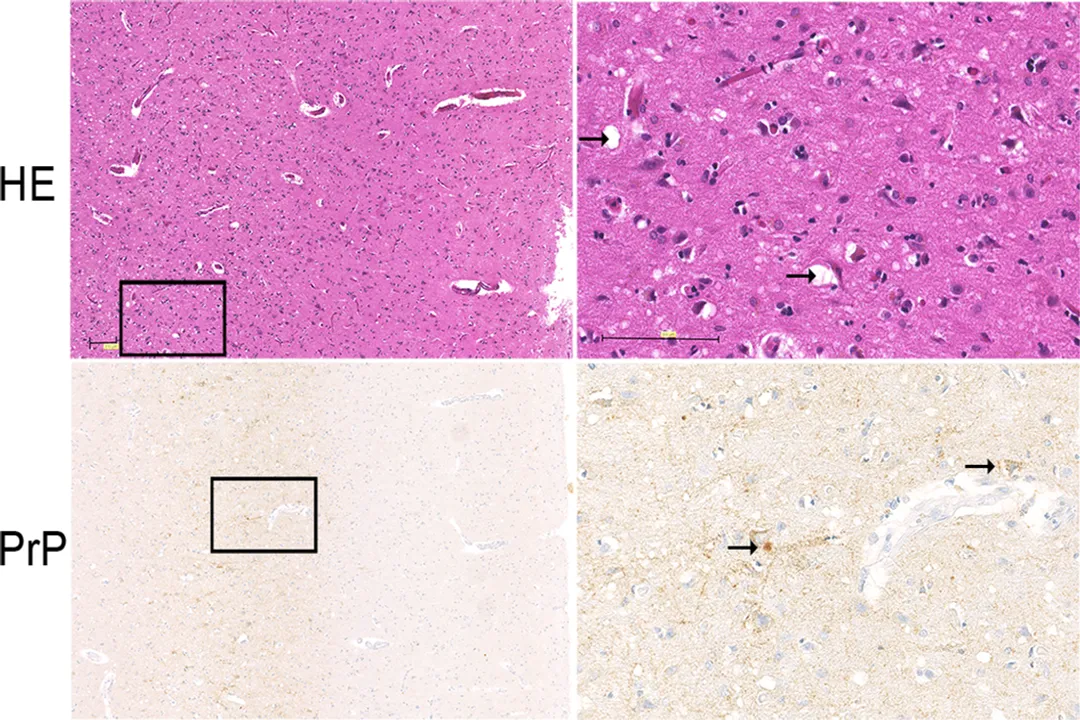
The relentless accumulation of these toxic, insoluble aggregates targets the most complex tissue in the body: the central nervous system. [1][8] The presence of large deposits and the failure of cellular processes lead to widespread loss of neurons. [3][4] This neuronal death is not a slow, quiet event; it results in significant structural damage to the brain matter. [1]
A defining macroscopic feature of nearly all prion diseases is the creation of vacuolae—small, empty spaces—within the gray matter of the brain, which leads to a spongy or sieve-like appearance. [1][3][4][8] This condition is scientifically termed spongiform encephalopathy. [8] This pathological hallmark reflects the extensive tissue destruction that has occurred. [1] Clinically, this progressive loss of functional brain tissue manifests as debilitating and incurable neurological symptoms, including dementia, ataxia (loss of coordination), and eventual death. [8]
To grasp the extent of the threat, consider the challenges in decontamination. Standard autoclaving procedures, which rely on high heat and pressure to denature proteins and kill microbes, are often insufficient to completely eliminate the infectious potential of . [2] This recalcitrance stems directly from the protein's highly stable beta-sheet conformation; it simply does not unfold under typical sterilization conditions. [7] This is why hospitals must use harsh chemical treatments, such as strong concentrations of sodium hydroxide or prolonged exposure to high-temperature steam, to safely process instruments that may have come into contact with infected tissue, a requirement far stricter than that for most bacterial or viral pathogens. [1][2]
# Disease Spectrum
Prion diseases appear across the animal kingdom and affect humans, presenting in three broad etiological categories: sporadic, inherited, and acquired. [8]
# Human Conditions
The most common form in humans is Creutzfeldt-Jakob disease (CJD), which accounts for about 85% of all cases and is classified as sporadic, meaning it occurs randomly without an identifiable external cause or genetic link. [8][3] Other human prion diseases include Fatal Familial Insomnia (FFI), which primarily impairs the sleep-wake cycle, and Kuru. [3] Kuru is a particularly striking historical example, linked to ritualistic cannibalism among the Fore people of Papua New Guinea, where the disease was transmitted by consuming infected human brain tissue during funerary practices. [2][8]
# Animal Ailments
In the animal world, the diseases are often named after the most noticeable clinical sign, such as wasting or tremors. [9] Scrapie is a long-recognized disease affecting sheep and goats. [4] Bovine Spongiform Encephalopathy (BSE), widely known as "mad cow disease," affected cattle, leading to significant agricultural concern when it was discovered that consuming BSE-infected beef products could transmit the disease to humans in the form of a variant CJD (). [1][2] A growing concern in North America is Chronic Wasting Disease (CWD), which affects wild cervids like deer, elk, and moose. [9]
The table below summarizes key recognized prion diseases and their typical hosts or origins:
| Disease | Primary Host | Etiology Category | Key Characteristic |
|---|---|---|---|
| CJD | Humans | Sporadic (most common) | Rapidly progressive dementia [3] |
| vCJD | Humans | Acquired (via BSE) | Affects younger individuals than sporadic CJD [8] |
| Kuru | Humans | Acquired (via ingestion) | Associated with funerary brain consumption [2] |
| Scrapie | Sheep/Goats | Natural (often sporadic/genetic) | Intense itching and muscle spasms [4] |
| BSE | Cattle | Acquired (via feed contamination) | Affects nervous tissue; led to dietary restrictions [1] |
| CWD | Deer/Elk/Moose | Natural (sporadic/environmental) | Progressive weight loss and neurological deficits [9] |
[^This table combines information on transmission, host, and clinical signs mentioned across several sources, offering a structural comparison that highlights the diversity of prion pathology across species [1][2][3][4][9].]
# Inheritance and Acquisition
While the sporadic form of CJD dominates human cases, the mechanism can also be genetically predetermined. Inherited prion diseases arise from mutations in the gene, the specific segment of DNA that carries the instructions for building the normal protein. [8] These mutations make the resulting molecule inherently unstable, predisposing it to spontaneously misfold into the form over time, leading to conditions like inherited CJD or FFI. [8]
Acquired prion diseases, although the rarest, demonstrate the infectious nature most clearly. [8] Transmission occurs when an individual comes into contact with, or consumes, infectious material. [2] Besides the historical example of Kuru, medical settings have posed risks. Because prions resist standard heat and chemical sterilization, there have been rare instances of transmission through contaminated surgical instruments, corneal grafts, dura mater grafts, or human growth hormone derived from infected pituitary glands. [1][2][8] These highly specific, iatrogenic (medically induced) routes underscore why vigilance in handling tissues from affected individuals is paramount for public health officials. [2]

# Environmental Factors and Spread
The environment can also become a reservoir for infectious prions, particularly in wildlife populations. Chronic Wasting Disease (CWD) illustrates this environmental persistence. [9] In areas where CWD is present, the infectious prions can contaminate the soil and water from infected animals that have died. [9] This presents a long-term contamination risk because the is incredibly resilient and can remain infectious in the environment for extended periods, posing a continuous threat to other susceptible animals that graze or inhabit that area. [9]
This environmental persistence is a concept that distinguishes prion pathology from most other infectious diseases. When a virus or bacterium contaminates a surface, standard cleaning agents and a bit of time usually neutralize the threat. [7] Prions, however, survive chemical inactivation and denaturation due to their unique structure. [4] This suggests that the breakdown mechanism in the environment is extremely slow, likely requiring intense UV radiation or prolonged exposure to corrosive chemicals rather than simple biological degradation. [5] This environmental stability means that proper disposal of infected carcasses and decontamination of premises is a major logistical challenge in managing outbreaks like CWD or BSE. [9]
# Progression of Symptoms
Regardless of whether the initial cause was sporadic, genetic, or acquired, the progression once the misfolded proteins start accumulating in the brain follows a similar, devastating trajectory. [8] The neurological symptoms often begin subtly. For example, in sporadic CJD, early signs might include vague changes in vision, balance issues, or mood disturbances. [8] As the prion plaques expand and the spongiform damage worsens, the symptoms accelerate and become more severe. [1]
Patients typically progress toward significant cognitive decline, developing dementia that worsens rapidly over months. [8] Motor function is severely impaired, often resulting in involuntary muscle jerks called myoclonus, unsteadiness (ataxia), and eventual inability to walk or speak coherently. [3][8] The disease course is relentlessly progressive, leading to coma and death, usually within one year of symptom onset, though the timescale can vary depending on the specific prion strain and the patient's underlying genetic background. [8] The strain variation is important: different strains of can cause diseases with slightly different incubation periods and symptom profiles, even when they share the same basic structural component. [5] This heterogeneity further complicates research and diagnosis, as the precise molecular features dictating disease characteristics are still being mapped out. [5]
The speed at which these diseases progress, especially CJD, can be alarming when compared to other neurodegenerative conditions. For instance, Alzheimer's disease typically progresses over many years or even decades, allowing for a longer period of patient management and adaptation. [8] Prion diseases, however, often move from initial noticeable symptoms to complete incapacitation in a matter of months, placing immense strain on both the patient and the caregivers trying to navigate the aggressive neurological decline. [8] This rapid fatality rate is a direct consequence of the immediate and widespread toxicity caused by the protein aggregation cascade described earlier. [7]
# Diagnostic Challenges
Diagnosing a prion disease while a patient is alive presents significant hurdles because the defining feature—the massive accumulation of —is difficult to measure directly in living tissue. [5] Early diagnosis often relies on ruling out other causes of rapidly progressive dementia. [8]
Initial investigations usually involve imaging tests like MRI, which can reveal the characteristic changes in brain structure associated with spongiform degeneration, particularly hyperintensity in specific brain regions. [8] Electroencephalography (EEG) may show distinctive, generalized periodic sharp wave complexes in CJD, although this finding is not always present. [8] Crucially, definitive diagnosis often requires examining cerebrospinal fluid (CSF) for biomarkers, such as elevated levels of 14-3-3 protein, which is released when neurons are rapidly dying. [8] However, the gold standard for definitive confirmation has historically been the post-mortem examination of brain tissue. [5] Recent advancements are making headway in detecting the misfolded prions directly in the CSF or even blood, utilizing highly sensitive techniques that can amplify minute amounts of the abnormal protein, offering hope for earlier in vivo diagnosis in the future. [5] This ongoing development represents a critical area of scientific focus, as effective treatments cannot be implemented without timely and accurate identification of the disease process. [7]
#Videos
2-Minute Neuroscience: Prion Diseases - YouTube
Related Questions
#Citations
Prion Diseases | Johns Hopkins Medicine
About Prion Diseases - CDC
Prion Disease: What It Is, Types, Causes, Symptoms & Treatment
Prion - Wikipedia
Cellular and Molecular Mechanisms of Prion Disease - PMC
2-Minute Neuroscience: Prion Diseases - YouTube
When Proteins Become Infectious: Understanding Prion Disease ...
Overview of Prion Diseases - Neurologic Disorders - MSD Manuals
What are Prions? - Virginia Department of Wildlife Resources