What signals initiate apoptosis?
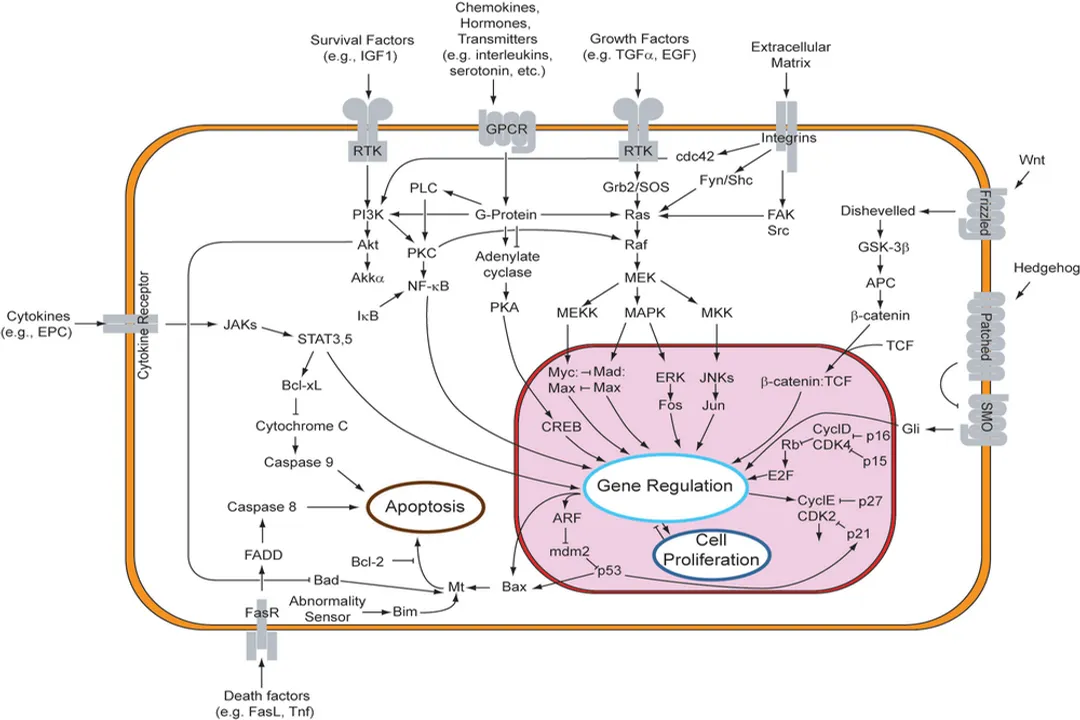
Programmed cell death, or apoptosis, is a fundamental biological process ensuring tissue homeostasis and removing damaged or unwanted cells without causing inflammation, a critical distinction from messy cell disintegration known as necrosis. [3][7] This controlled dismantling is essential for embryonic development, immune system regulation, and safeguarding against diseases like cancer. [3][6] The initiation of this cascade is not random; it requires specific, precise molecular signals that tell the cell it is time to self-destruct in an orderly fashion. These initiation signals converge upon central execution machinery, primarily a family of proteases called caspases, which then dismantle the cell from the inside out. [1][8]
# Cell Demarcation
Before diving into the triggers, it’s useful to position apoptosis relative to other cell fate decisions. Unlike necrosis, which is typically a result of acute injury—leading to cell swelling, rupture, and leakage of intracellular contents into the surrounding tissue (often causing inflammation)—apoptosis is an active, genetically controlled process. [3] During apoptosis, the cell shrinks, the chromatin condenses, and the cell membrane forms blebs, packaging the cellular contents into neat little packages called apoptotic bodies that phagocytic cells can easily clear. [3] The signals that initiate this process are generally categorized based on where they originate: from outside the cell or from within. [1][6]
# External Triggers
The extrinsic pathway of apoptosis is initiated by signals originating from external cues, often through specialized cell surface receptors known as death receptors. [4][8] These receptors belong to the tumor necrosis factor (TNF) receptor superfamily and are found on the cell surface. [4]
When an external signal binds to a death receptor, it acts as the "go" signal for cell suicide. [6] A classic example involves the interaction between a cell displaying a death ligand, such as Fas ligand (FasL) or TNF-alpha, and a receiving cell that expresses the corresponding receptor, like Fas (CD95) or TNFR1. [4]
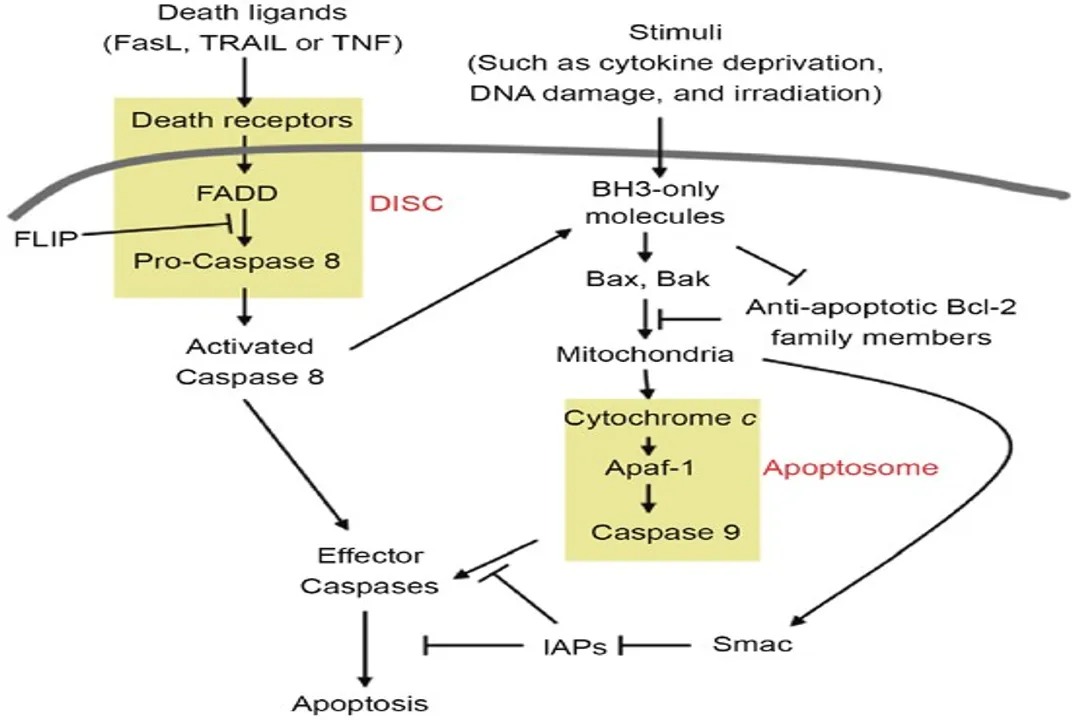
Upon ligand binding, the receptor trimerizes (three receptor units come together), which causes the recruitment of adapter proteins to the intracellular death domains of the receptors. [4] This assembly forms a complex often termed the Death-Inducing Signaling Complex (DISC). [4] Within the DISC, the initiator caspase, specifically procaspase-8, is recruited and activated through proximity-induced cleavage. [4] Once activated, caspase-8 can directly cleave and activate downstream executioner caspases, like caspase-3, immediately setting the demolition process in motion. [4] This pathway is critical for immune surveillance, for instance, in eliminating unwanted lymphocytes or virus-infected cells. [4]
# Internal Stressors
While external signals provide external control, many times the cell initiates apoptosis due to internal distress, activating the intrinsic pathway, sometimes referred to as the mitochondrial pathway. [1][6][8] This pathway responds to a variety of intracellular stresses that signal irreparable damage or unfavorable conditions. [8]
Key internal signals that activate this route include:
- Irreparable DNA damage: Often caused by radiation or chemical agents. [8]
- Oxidative stress: An imbalance between reactive oxygen species (ROS) production and the cell's ability to detoxify them. [6]
- Growth factor withdrawal: The lack of necessary survival signals from the cellular environment. [1][8]
- Endoplasmic Reticulum (ER) stress: Accumulation of unfolded or misfolded proteins within the ER. [1]
When these stresses reach a critical threshold, they trigger the permeabilization of the outer mitochondrial membrane. [1][6] This event is the hallmark of intrinsic pathway activation and represents a point of no return. [8] The permeabilization allows crucial pro-apoptotic proteins, which normally reside in the mitochondrial intermembrane space, to leak out into the cytoplasm. [1] The most critical of these is Cytochrome c. [1][6][8]
Once released, Cytochrome c binds to a protein called Apaf-1 (Apoptotic protease activating factor 1) in the presence of deoxyadenosine triphosphate (dATP). [8] This complex then oligomerizes to form a large structure known as the apoptosome. [1][8] The apoptosome acts as a scaffold to recruit and activate the initiator procaspase-9. [8] Activated caspase-9, in turn, initiates the caspase cascade by cleaving and activating the executioner caspases. [1][6]
# Crossover Mechanisms
It is worth noting that the extrinsic and intrinsic pathways are not entirely isolated; they possess mechanisms to cross-talk, which serves to amplify the death signal and ensure the process completes efficiently. [4][6]
In the extrinsic pathway, when caspase-8 is activated at the DISC, it doesn't only activate executioner caspases directly. [4] In many cell types, especially those with low levels of the protein BID, activated caspase-8 cleaves the pro-survival protein Bid (tBid), generating truncated Bid (tBid). [6] This truncated version is a powerful activator of the intrinsic pathway—it translocates to the mitochondria and promotes the release of Cytochrome c, thereby engaging the apoptosome and caspase-9 amplification loop. [4][6] This integration ensures that even a relatively weak initial external signal can lead to a strong, irreversible apoptotic commitment through the powerful mitochondrial route. [4]

# Regulatory Players
The decision to initiate apoptosis is tightly regulated by a family of proteins that monitor the health of the cell and govern the permeability of the mitochondrial membrane in the intrinsic pathway. [1][6] These regulators are part of the Bcl-2 family of proteins. [1][6][8]
This family can be generally divided into three functional groups, which determine whether the cell survives or dies:
- Anti-apoptotic proteins: These proteins, such as Bcl-2 and Bcl-XL, reside on the mitochondrial outer membrane and actively work to prevent permeability, thus inhibiting Cytochrome c release and keeping the cell alive. [1][6] They essentially stabilize the membrane. [8]
- Pro-apoptotic effectors: These are the executioners of the mitochondrial pathway, like Bax and Bak. [1][6][8] When activated, these proteins insert themselves into the outer mitochondrial membrane, forming pores that allow the leakage of pro-apoptotic factors like Cytochrome c. [1][8]
- Pro-apoptotic BH3-only proteins: These act as the sensors or mediators. [1][6] Proteins like Bim and Bad respond directly to the initial stress signals (like growth factor withdrawal or DNA damage). [6] They initiate the commitment to die by inhibiting the anti-apoptotic proteins (like Bcl-2) or by directly activating the effector proteins (like Bax/Bak). [1][6]
The balance between these opposing forces dictates the cell's fate. If the signals favoring death (the BH3-only proteins and activated effectors) overcome the survival signals (the anti-apoptotic Bcl-2 proteins), the mitochondria commit to releasing their contents, and apoptosis proceeds. [8] This regulatory balance is where many anti-cancer drugs aim to intervene, tipping the scales toward death in malignant cells that have otherwise learned to silence death signals. [2] A disruption in this balance, where anti-apoptotic signals dominate inappropriately, is a characteristic feature of many cancers. [2]
# The Execution Phase Signals
The true signal for dismantling the cell occurs when executioner caspases become active. [1] Once initiated, the cell has a cascade of self-destruction mechanisms underway, controlled by caspases-3, -6, and -7. [1] These are activated by initiator caspases (like caspase-8 or -9). [6] Their activation is itself a powerful signal, as they cleave hundreds of specific cellular substrates, leading directly to the morphological hallmarks of apoptosis. [8]
For example, executioner caspases cleave structural proteins of the cytoskeleton, causing the cell to change shape and shrink. [8] They also cleave the enzyme ICAD (Inhibitor of Caspase-Activated DNase). [1] When ICAD is cleaved, its inhibitory grip is released, allowing the DNase enzyme to enter the nucleus and begin systematically fragmenting the DNA into characteristic ladder-like patterns. [1][8] This specific DNA fragmentation pattern is a biochemical hallmark used in research to confirm that cell death occurred via apoptosis. [1]
# Signal Integration and Context
What is fascinating is how the cell weighs these different inputs. Consider a scenario where a cell receives a weak death receptor signal (extrinsic) but is simultaneously bathed in survival factors (strong anti-apoptotic signal intrinsically). In this tug-of-war, the cell’s survival mechanisms are likely to prevail because the intrinsic regulators are highly sensitive. [6] However, if that same cell experiences mild DNA damage, the activation of a BH3-only protein like PUMA might rapidly neutralize the protective Bcl-2 proteins, allowing the external death signal—which might otherwise have been ignored—to successfully trigger mitochondrial leakage and subsequent death.
This interplay suggests that the intensity and concurrency of signals matter as much as the presence of a single trigger. For a researcher observing cell death in a dish, distinguishing between a purely extrinsic or purely intrinsic signal can be tricky because most biological contexts involve crosstalk. If one is testing a compound intended to induce apoptosis, observing a reduction in Cytochrome c release following treatment, coupled with reduced caspase-9 activation but maintained caspase-8 activation, would strongly imply the compound is acting by strengthening the intrinsic signaling components, perhaps by directly neutralizing Bcl-2 proteins. [1][6]
A practical consideration for those working with cell models highlights the need to control for baseline stress. If a cell line is already under high ER stress due to suboptimal culturing conditions—perhaps too high a density or nutrient depletion—its intrinsic pathway might be primed for activation. [1] Introducing an extrinsic stimulus might then cause massive, premature cell death that wouldn't occur in a perfectly healthy cell culture. Thus, maintaining optimal growth factor concentration and nutrient availability is an indirect, but crucial, pre-emptive signal to inhibit apoptosis initiation, even when studying external death pathways. [8] Think of it this way: a healthy cell has a high threshold for internal breakdown signals; an already stressed cell has a lowered threshold, meaning it takes less additional bad news to initiate the cascade. [6]
# Receptors and Adaptors Summary
To summarize the initial inputs, the primary signals converge on a few key molecules that transduce the message across the cell membrane or within the cytosol.
| Pathway | Initial Signal Source | Key Receptor/Sensor | Initial Downstream Complex |
|---|---|---|---|
| Extrinsic | External Ligands (e.g., FasL, TNF-a) | Death Receptors (Fas, TNFR1) | DISC (Death-Inducing Signaling Complex) [4] |
| Intrinsic | Internal Stress (e.g., DNA damage, ROS) | Bcl-2 Family Proteins (e.g., Bad, Bim) | Apoptosome (via Cytochrome c) [8] |
The activation of caspase-8 by the DISC in the extrinsic path, or caspase-9 by the apoptosome in the intrinsic path, represents the commitment point where the initiating signal transitions into the irreversible execution signal. [1][8] Furthermore, the ability of activated caspase-8 to cleave Bid, thereby activating the intrinsic machinery, means that any strong extrinsic signal can effectively become an intrinsic one, ensuring signal amplification. [4] This redundancy and interlinking between the two initiation routes are central to the fidelity of programmed cell death. [6]
# Regulatory Failure Signals
When apoptosis fails to initiate correctly, the consequence is often uncontrolled cell accumulation, a hallmark of cancer. [2] Conversely, excessive, inappropriate initiation of apoptosis contributes to degenerative diseases. [7] For example, a mutation that causes a pro-apoptotic BH3-only protein, like Bim, to be continuously expressed at high levels, regardless of external survival cues, will drive cells into unwarranted death. [6] On the flip side, the overexpression of anti-apoptotic proteins, such as Bcl-2, allows cells that should be eliminated (due to mutations or damage) to survive and proliferate indefinitely. [2] Understanding which specific initiating signals are blocked or amplified in a diseased state is key to developing targeted therapies that aim to restore the cell's ability to respond correctly to death cues. [2]
Related Questions
#Citations
Apoptosis Signaling Pathway: Stages, Types and Key Molecules
Apoptosis signaling pathways and lymphocyte homeostasis - Nature
Apoptosis - Wikipedia
Signaling Pathway - Novus Biologicals
Intrinsic and Extrinsic Pathways of Apoptosis - Thermo Fisher Scientific
Apoptosis Signal Transduction Pathway - Bio-Techne
Apoptosis (Programmed Cell Death) - Cleveland Clinic
Apoptosis Signaling Pathways - R&D Systems
Apoptosis signaling - PubMed