What mechanisms underlie pain perception?
The very essence of pain, that urgent signal alerting us to danger or damage, is a profound biological process built upon layers of cellular detection and complex brain processing. It is far more than a simple electrical wire running from a source of injury straight to our consciousness; it is an intricate system that involves specialized sensors, chemical messengers, and remarkable neural plasticity in both the periphery and the central nervous system (CNS). [5]
For centuries, thinkers tried to pin down what pain was. Early concepts, dating back to ancient Greek medicine, often linked pain to imbalances in bodily elements like bile and phlegm, essentially viewing it as a product of the brain, or an emotion, rather than a pure physical sensation. The Scientific Revolution brought more mechanistic models. René Descartes described a system where a stimulus tugged on a nerve "tubule," opening a gate to allow "animal spirits" to flow to the brain, causing the perception. This began the distinction between the physical event (nociception) and the subjective experience (pain).
# Signaling Stages
The modern view breaks down the journey of a pain signal into distinct, albeit interconnected, stages: transduction, transmission, and perception, which is further refined by modulation. [4]
# Transduction At the Edge
Pain perception begins at the endings of specialized primary sensory neurons called nociceptors. [5][6] These are the body’s sensors for stimuli deemed noxious—that is, intense enough to cause actual or potential tissue damage. [5] Nociceptors are highly heterogeneous, differing in the receptors they express and the speed at which they conduct signals. [5][6]
Nociceptors primarily come in two types based on their fiber characteristics:
- A-fiber nociceptors: These have lightly myelinated axons, allowing for faster conduction, and typically mediate the initial, fast, pricking quality of pain. [5]
- C-fibers: These have unmyelinated axons, leading to slower conduction, and are responsible for the slower, burning quality of pain. [5] C-fibers make up about 70% of all nociceptors. [5]
The conversion of a noxious stimulus (heat, cold, pressure, or chemical irritant) into an electrical signal is called transduction. [4] This happens via specific ion channels located at the peripheral nerve endings. [5][6]
For noxious heat, the Transient Receptor Potential Vanilloid 1 () channel is a key player. Activation of converts thermal energy into an electrical signal sent toward the CNS. [5] However, research indicates that other heat sensors besides must exist, as deleting in mice only partially reduces noxious heat sensitivity. [5]
Noxious cold detection also relies on specific channels. The TRPM8 channel is known to detect innocuous cooling, but it also contributes to sensing noxious cold, although its role is still being defined across species. [5] The TRPA1 channel is another candidate for cold sensation, often implicated in the burning sensation associated with irritants like mustard oil, which also activates -expressing neurons. [5]
Mechanical stimuli, like intense pressure, remain the most elusive in terms of precise molecular transduction, though Acid-Sensing Ion Channels (ASICs) are thought to play some part. [5]
# Transmission and Wiring
Once a nociceptor is depolarized enough to fire an action potential, this electrical signal travels along the axon to the spinal cord. [4][5] The central terminals of these primary afferents synapse onto second-order neurons in the dorsal horn of the spinal cord. [5][6]
The historical debate centered on whether these pathways were entirely dedicated or if the pattern of firing determined the sensation. Modern understanding suggests a hybrid approach: the peripheral encoding often does follow a labeled line fashion, meaning specific fiber types signal specific qualities like heat or cold. However, the resulting information is processed centrally in a much more complex manner.
Nociceptors are not all the same even within the A-fiber/C-fiber distinction. For instance, C-fibers can be categorized based on their expression of the carbohydrate group: [5][6]
- -negative C-fibers: These express receptors and project to the superficial laminae ( and outer ) of the dorsal horn, synapsing on neurons that project to higher pain centers. [5] They appear particularly responsive to noxious heat and chemicals. [5][6]
- -positive C-fibers: These project to a different area (inner lamina ) and synapse primarily on local spinal interneurons. [5]
This division in spinal termination—one pathway leading directly "up" to the brain's sensory and affective centers, the other connecting locally—highlights how different input types may generate distinct aspects of the pain experience. [5]
# Central Processing and Plasticity

The real shift from an immediate warning signal to a persistent problem occurs centrally, involving neuronal plasticity—the nervous system’s ability to change its structure and function. [5] This plasticity is a core mechanism underlying chronic pain, often categorized as central sensitization.
# Spinal Changes: Priming and Summation
Peripheral nerve activation can lead to both short-term and long-term changes in the CNS. [5] Two related phenomena at the spinal level drive this change:
- Wind-up: This occurs from repetitive low-frequency input to -fibers. It results in temporal summation, where the neuron’s response grows larger with each subsequent stimulus, leading to hypersensitivity characterized by lowered thresholds to maintain the response. [1]
- Hyperalgesic Priming: This is a long-term alteration where a peripheral nociceptor becomes more excitable due to a prior injury or priming event. [1] This involves local changes in the afferent nerve ending, potentially through localized protein synthesis at the synapse, driven by mediators like Nerve Growth Factor () and Interleukin-6 (). [1]
When these processes are established, the spinal cord begins to amplify ascending signals, contributing to the persistent nature of chronic pain. [1]
# Brain Remodeling
If peripheral nociceptive input is excessive or prolonged, it results in functional and structural changes in brain regions associated with pain perception, emotion, and cognition. [1][3] This engagement of brain areas critical for cognitive/emotional assessments is what distinguishes the network for chronic pain from that of acute pain. [3]
Key brain areas implicated in this remodeling include:
- The Anterior Cingulate Cortex () and Insular Cortex (), which process the affective (unpleasantness) and cognitive-evaluative components of pain. [1] Imaging studies show alterations in functional connectivity and gray matter density in these areas in chronic pain patients. [1]
- The Amygdala (CeA), which is vital for pain regulation and the emotional response to pain; repetitive activation can cause remodeling in this area. [1]
The complexity arises because chronic pain is often not just an over-active ascending signal but also involves a failure of the descending control system. [3] Normally, pathways originating in the Periaqueductal Gray () and Rostral Ventromedial Medulla () project to inhibitory interneurons in the spinal cord, effectively turning the pain signal off. Central sensitization can involve the decreased function of these inhibitory interneurons, allowing more signal through, even without a strong peripheral drive. [1]
# The Role of Stress and Neuroimmune Crosstalk
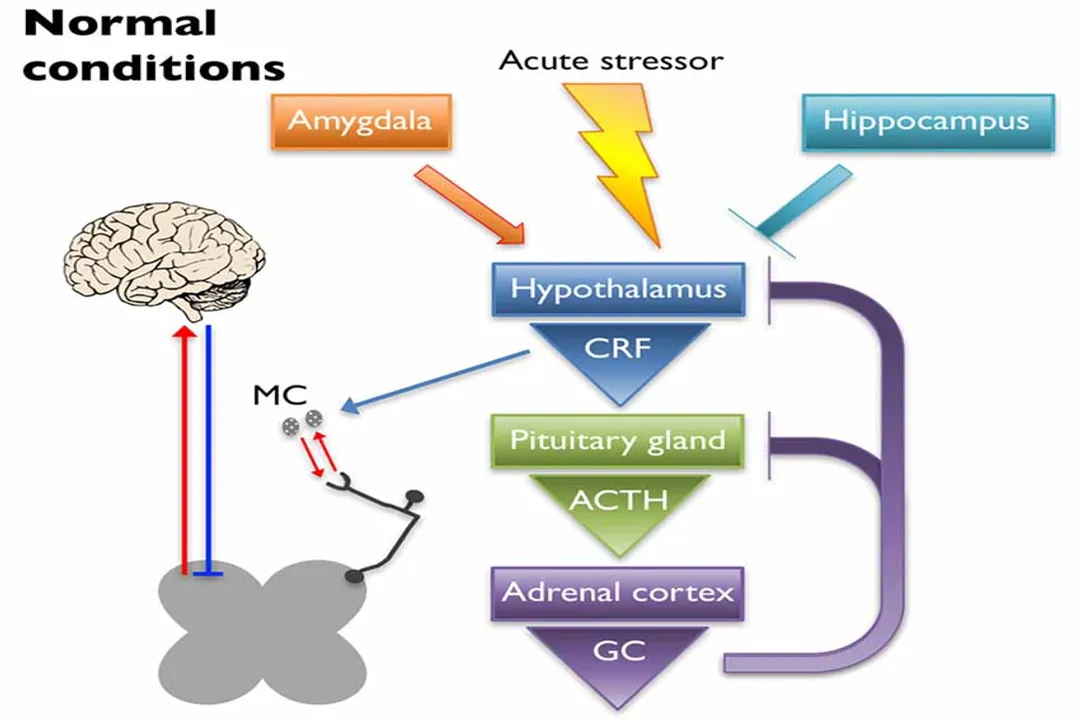
A significant factor complicating the mechanisms of chronic pain, particularly in conditions like fibromyalgia and chronic pelvic pain syndromes, is the dysregulation of the Hypothalamic-Pituitary-Adrenal () axis—the body's primary stress response system. [1]
When the axis is chronically altered, often due to early life stress or trauma, it can lead to abnormal production of cortisol and downstream effects that increase nociceptive tone peripherally. [1] Corticotropin-releasing factor (), released during stress activation, can peripherally activate Mast Cells (). [1]
are immune cells packed with inflammatory mediators like histamine and proteases, and they are often found right next to sensory nerve endings. [1] When activated by or mast cell proteases (), release substances like and (). [1] These mediators then directly sensitize nearby nociceptors by interacting with channels like and , effectively lowering the pain threshold in the tissue. [1] This axis dysregulation, through this neuroimmune link, can contribute to the peripheral sensitization that feeds into central changes. [1]
# Theoretical Evolution
This multifaceted understanding required several theoretical shifts. The historical move from Specificity Theory (dedicated pain wires) to the Intensity/Pattern Theories (pain from overwhelming or patterned signals) eventually resolved into the Gate Control Theory.
The Gate Control Theory proposed the spinal cord acts as a gate modulated by both large-diameter (touch/non-painful) fibers closing it and small-diameter (pain/C-fibers) fibers opening it, with top-down influence from the brain also modulating the mechanism. [4] While the precise neural architecture described in the original 1965 model has been refined, its core concept—that non-painful input and central control can suppress pain transmission—remains highly influential.
Contemporary pain research recognizes that pain is multidimensional, involving sensory-discriminative, affective-motivational, and cognitive-evaluative components, which all interact. Pain intensity might be high, but a person’s distress (affective component) can be modulated separately by their cognitive state, such as through expectation or belief. For example, hypnosis can modulate the unpleasantness of pain without changing the perceived intensity.
| Theory | Core Tenet | Relevance Today |
|---|---|---|
| Specificity Theory | Dedicated receptors and fibers exist for each sensation, including pain. | Partially true at the periphery; specific nociceptors are specialized for heat, cold, etc.. [5][6] |
| Intensity/Pattern Theory | Pain arises from the intensity or pattern of nerve firing, not unique receptors. | Addresses how non-nociceptive fibers, when overstimulated, can contribute to pain perception. |
| Gate Control Theory | Spinal cord "gate" modulates signal flow based on A-fiber, C-fiber, and descending input. | Provides the neural basis for understanding how non-painful input inhibits pain. [4] |

# Chronic States and Opioid Resistance
The mechanisms shift when pain becomes neuropathic—caused by actual lesion or disease of the somatosensory nervous system—or transitions to a chronic state. A striking difference in neuropathic pain is its frequent insensitivity to opioid analgesics like morphine. [6] This resistance may stem from decreased -opioid receptor expression on the affected neurons, or it can be due to increased counter-modulation, such as by the neuropeptide cholecystokinin (), which reduces morphine's inhibitory effects. [6] Consequently, neuropathic pain is often managed with agents that modulate neurotransmitters differently, like antidepressants and anti-epileptics. [6]
Chronic pain, or chronification, involves neuroplastic changes that can become self-perpetuating, sometimes leading to the perception of pain without continued noxious input—the hallmark of central sensitization. [1] This results in allodynia (pain from a non-noxious stimulus) and hyperalgesia (increased pain from a noxious stimulus) in widespread areas. [1] Conditions like fibromyalgia and migraine frequently exhibit this central sensitization, evidenced by abnormal brain structure and function in those with allodynia. [1]
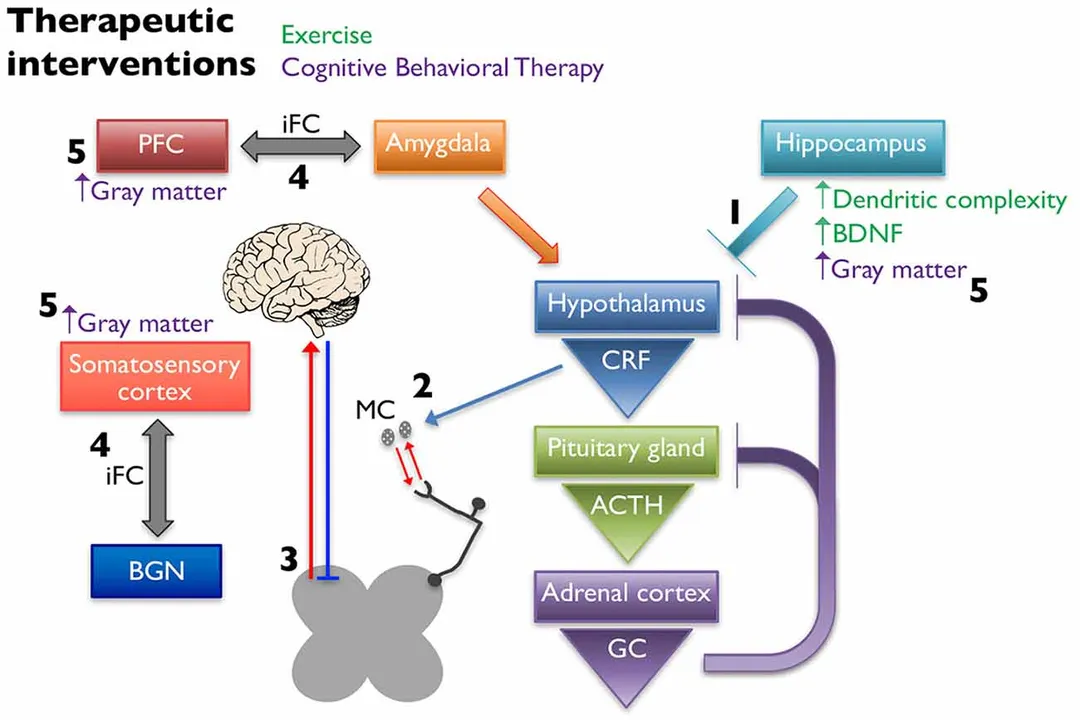
When considering the axis link, it’s fascinating to note that stress-induced changes can amplify peripheral input via activation, leading to hyperalgesic priming, while simultaneously affecting descending inhibition from the brain. [1] This suggests a two-pronged attack that makes breaking the cycle of chronic pain so difficult. A practical implication here is that for patients with a history of significant stress or trauma, directly addressing the axis and stress reactivity through therapies like Cognitive Behavioral Therapy () may be just as critical as managing peripheral tissue input, because the underlying neuroimmune environment is already sensitized. [1]
# Altering the Experience
Because the entire process is so complex and involves brain regions for emotion and cognition, non-pharmacological approaches that target these higher centers show promise, especially where traditional medications fail or cause side effects. [1]
# Physical Activity
Exercise, when appropriately dosed, can exert analgesic effects. This is partly mediated by influencing the axis, normalizing levels of stress-related markers in the hippocampus, and potentially restoring inhibitory control. [1] Exercise may also improve Conditioned Pain Modulation (), the body’s innate "pain-inhibits-pain" system, which is often defective in chronic pain patients. [1] The mechanism may involve increasing endogenous opioids in the CNS, or by balancing excitatory () and inhibitory () neurotransmitters. [1] Crucially, intensity matters; low-to-moderate intensity aerobic training often helps fibromyalgia patients, whereas overly strenuous exercise can sometimes increase pain, suggesting a narrow therapeutic window. [1]
# Cognitive Reframing
Cognitive Behavioral Therapy () directly targets the cognitive-evaluative dimension of pain. It teaches coping mechanisms to reduce the emotional impact and stress response associated with pain, which, in turn, is hypothesized to reduce axis activation and its negative downstream effects. [1] Neuroimaging has revealed that successful can actually induce structural changes, such as increases in gray matter volume in sensory and motor brain areas, and alter functional connectivity between regions involved in pain management, like the prefrontal cortex and amygdala. [1]
The shift in understanding has moved from viewing pain as a simple sensory event to recognizing it as a state of the nervous system—one that is shaped by genetics, past experience, current emotional state, and ongoing neuroplasticity. [3] The integration of these multiple pathways—from the channel in a peripheral nerve ending to the functional connectivity of the —is the key to deciphering the mechanisms that underlie an individual's unique experience of pain. [3][5]
# The Interplay of Mechanisms
A key insight derived from comparing these findings is that the mechanisms causing persistent pain are rarely isolated. For example, in centralized pain syndromes, the dysregulation of the axis (a stress/neuroendocrine factor) is mechanistically linked to central sensitization (a neuronal plasticity factor) via the activation of mast cells in the periphery. [1] This means that the bottom-up process of constant noxious input sensitizing the spinal cord and brain is often fueled by top-down influences originating from stress responses. [1] For example, rodent models show that early life stress leads to widespread pain, regardless of whether the immediate effect on the HPA axis was hyper- or hypocortisolism; the dysregulation itself seems to lead to the same adverse outcome of heightened sensitivity. [1]
This convergence strongly suggests that effective management cannot target just one area. If an individual is experiencing widespread pain, the CNS has already adapted. [5] Therefore, treatment protocols that combine elements to calm the periphery, restore descending inhibition, and retrain the brain's interpretation of the signal—perhaps through tailored exercise and —offer the best prospect for moving closer to the vision of a world less burdened by chronic suffering. [1][4]
Related Questions
#Citations
The Science Behind Pain: Mechanism of Pain Perception
Human Brain Pain Perception & Regulation: Health & Disease
Mechanisms of pain - PNAS
Nociceptors: the sensors of the pain pathway - JCI
Theories of pain: from specificity to gate control
Mechanisms of Centralized Pain & Therapeutic Interventions
Pathophysiology | Grünenthal